前処理で重複する分子データを効率的に除去
こんにちは!最近、SMILESを用いた機械学習をやっていまして、その際にデータセットの構造式が重複しているケースがありました。
機械学習や化学データ解析において、データセット内の重複分子データを正確に除去することは重要です。特に、SMILES表記(Simplified Molecular Input Line Entry System)を扱う際、同一分子でも異なるSMILES表記が存在するため、それらを適切に処理する必要があります。
この記事では、RDKitを活用してSMILES表記の重複分子を簡単に判別する方法を解説します。実際のコード例とともに詳しく見ていきましょう!
同一分子を表す異なるSMILES表記の例
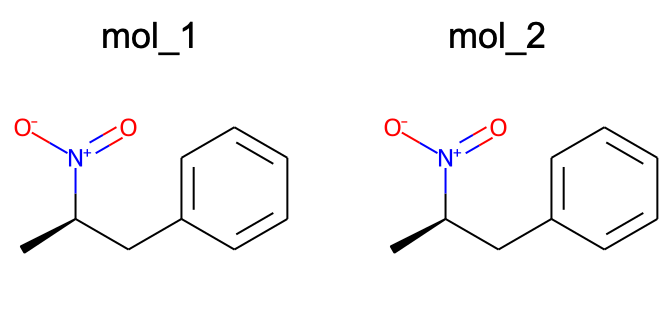
(R)-2-ニトロ-1-フェニルプロパンを表す2通りのSMILESは以下の通りです。いずれも同じ分子を表記するためのSMILESです。
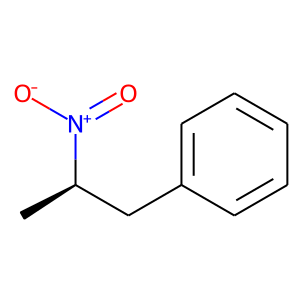
SMILES例 mol_1: C[C@H](CC1=CC=CC=C1)[N+](=O)[O-]
SMILES例 mol_2: C[C@H](Cc1ccccc1)[N+](=O)[O-]
これらのSMILESは異なりますが、同一の分子を表しています。それを確認するために、RDKitを使用して構造式を表示します。同じ構造式であることがわかりますね!
表示するためのコードは以下の通りです。
from rdkit import Chem
from rdkit.Chem import Draw
smiles_1 = "C[C@H](CC1=CC=CC=C1)[N+](=O)[O-]"
smiles_2 = "C[C@H](Cc1ccccc1)[N+](=O)[O-]"
mol_1 = Chem.MolFromSmiles(smiles_1)
mol_2 = Chem.MolFromSmiles(smiles_2)
# 分子を表示
Draw.MolToImage(mol_1)
Draw.MolToImage(mol_2)重複データを判別するコード例
もし、機械学習をしていて、データセットに本来は同じ分子を示すにも関わらず異なるSMILESを持つmol_1とmol_2があった場合、特徴量エンジニアリングの際にどちらかを消去することで過学習を防ぐ必要があります。
データセットに同一分子を示す異なるSMILES表記が含まれている場合、以下のコードを使用して重複を除去できます。RDKitのMolToSmiles関数を使用して標準化されたSMILESに変換することで、同一性を判別します。
from rdkit import Chem
smiles_1 = "C[C@H](CC1=CC=CC=C1)[N+](=O)[O-]"
smiles_2 = "C[C@H](Cc1ccccc1)[N+](=O)[O-]"
mol_1 = Chem.MolFromSmiles(smiles_1)
mol_2 = Chem.MolFromSmiles(smiles_2)
# 標準化されたSMILESに変換
mol_1_updated = Chem.MolToSmiles(mol_1)
mol_2_updated = Chem.MolToSmiles(mol_2)
print(mol_1_updated) # 出力: 'C[C@H](Cc1ccccc1)[N+](=O)[O-]'
print(mol_2_updated) # 出力: 'C[C@H](Cc1ccccc1)[N+](=O)[O-]'これにより、異なるSMILES表記でも同一の化合物であることを確認できます。
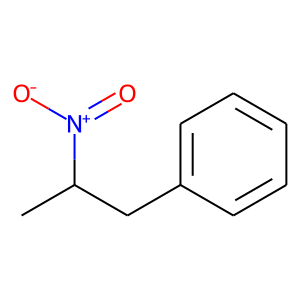
キラリティを無視した重複判別
場合によっては、キラリティを無視して同一分子かどうかを判別したい場合があります。RDKitのRemoveStereochemistryを使用することで、立体情報を削除できます。
from rdkit import Chem
from rdkit.Chem import Draw
smiles_1 = "C[C@H](CC1=CC=CC=C1)[N+](=O)[O-]"
smiles_2 = "C[C@H](Cc1ccccc1)[N+](=O)[O-]"
mol_1 = Chem.MolFromSmiles(smiles_1)
mol_2 = Chem.MolFromSmiles(smiles_2)
mol_1_RS = Chem.RemoveStereochemistry(mol_1)
mol_2_RS = Chem.RemoveStereochemistry(mol_2)
mol_1_RS = Chem.MolToSmiles(mol_1) # 'CC(Cc1ccccc1)[N+](=O)[O-]'
mol_2_RS = Chem.MolToSmiles(mol_2) # 'CC(Cc1ccccc1)[N+](=O)[O-]'mol_1_RSとmol_2_RSを構造式で表示するとどちらも以下のようになります。キラリティは判別できませんが、同じ分子かを判別できるのは便利ですね!
使用したコードの解説
MolFromSmiles
SMILES文字列から分子オブジェクト(Mol)を生成します。
from rdkit import Chem
mol = Chem.MolFromSmiles('CCO') # エタノールのSMILES表記MolToSmiles
分子オブジェクト(Mol)からカノニカルSMILES(標準化されたSMILES)文字列を生成します。
from rdkit import Chem
smiles = Chem.MolToSmiles(mol)
print(smiles) # 出力: 'CCO'RemoveStereochemistry
分子オブジェクトから全ての立体化学情報を削除します。
from rdkit import Chem
from rdkit.Chem import rdmolops
mol = Chem.MolFromSmiles('Cl[C@H](F)Br')
rdmolops.RemoveStereochemistry(mol)
smiles = Chem.MolToSmiles(mol)
print(smiles) # 出力: 'ClC(F)Br'まとめ
RDKitを使用することで、異なるSMILES表記の分子が同一であるかを簡単に判別できます。特に、機械学習の前処理で重複データを除去する際に非常に便利です。また、キラリティ情報の有無に応じた柔軟な処理も可能です。
SMILESを扱う機会がある方は、ぜひこの記事で紹介した方法を活用してみてください!


コメント